
ISO 13485 implementation: Mandatory documents and records
Preparation of the QMS documentation is based on the organizational needs, and it is crucial for an efficient QMS. Besides, correctly organized documentation will make operations much easier.
The purpose of QMS documentation is to provide a clear framework for the company’s operations and a better understanding of the QMS, and finally to show the evidence of accomplishing the company’s goals and objectives.
This is the list of documents and records needed for compliance with ISO 13485:2016:
4.1. General requirements
- Clause 4.1.1: Roles undertaken by the organization under applicable regulatory requirements
- Clause 4.1.6: Procedure and records for the validation of the application of computer software
- Clause 4.2.2: Quality Manual
- Clause 4.2.3: Medical device file
- Clause 4.2.4: Procedure for document control
- Clause 4.2.5: Procedure for record control
5. Management responsibility
- Clause 5.3: Quality policy
- Clause 5.4.1: Quality objectives
- Clause 5.5.1: Responsibilities and authorities
- Clause 5.6.1: Procedure and records for management review
6. Resource management
- Clause 6.2: Procedure for training
- Clause 6.3: Requirements for infrastructure and maintenance activities
- Clause 6.4.1: Requirements for a work environment
- Clause 6.4.2: Arrangements for control of contaminated or potentially contaminated product
7. Product realization
- Clause 7.1: Process for risk management in product realization
- Clause 7.1: Outputs of product realization planning
- Clause 7.2.2: Records of the results of the customer requirements review and actions arising from it
- Clause 7.2.3: Arrangements for communication with customers
- Clause 7.3.1: Procedure for design and development
- Clause 7.3.2: Design and development planning
- Clause 7.3.4: Design and development outputs
- Clause 7.3.5: Records of design and development review
- Clause 7.3.6: Design verification plans, results, and conclusions
- Clause 7.3.7: Design validation plans, results, and conclusions
- Clause 7.3.8: Procedure for transfer of design and development outputs to manufacturing
- Clause 7.3.9: Procedure and records for control of design and development changes
- Clause 7.3.10: Design and development file
- Clause 7.4.1: Procedure for purchasing
- Clause 7.4.1: Criteria and records for evaluation and selection of suppliers
- Clause 7.4.3: Record of verification of purchased product
- Clause 7.5.1: Record for each medical device or batch that provides traceability
- Clause 7.5.2: Requirements for the cleanliness of the product
- Clause 7.5.3: Requirements for medical device installation and acceptance criteria for verification of installation
- Clause 7.5.3: Records for medical device installation and verification of installation
- Clause 7.5.4: Procedure and records for servicing of the medical device
- Clause 7.5.5: Records of sterilization process
- Clause 7.5.6: Procedure and records of production and service provision process validation
- Clause 7.5.7: Procedure and records for validation of process for sterilization and sterile barriers systems
- Clause 7.5.8: Procedure for product identification
- Clause 7.5.9.1: Procedure for traceability
- Clause 7.5.9.2: Records of traceability and name and address of the shipping package consignee
- Clause 7.5.10: Report on changes on customer property
- Clause 7.5.11: Procedure for preserving the conformity of product
- Clause 7.6: Procedure for monitoring and measuring
- Clause 7.6: Record of calibration
- Clause 7.6: Procedure and records for validation of the application of computer software used for monitoring and measuring
8. Measurement, analysis, and improvement
- Clause 8.2.1: Procedure for customer feedback
- Clause 8.2.2: Procedure and records for complaint handling
- Clause 8.2.3: Records of reporting to regulatory authorities
- Clause 8.2.4: Procedure for internal audit
- Clause 8.2.4: Records of audits and their results
- Clause 8.2.6: Identity of the person authorizing release of product
- Clause 8.3.1: Procedure and record of control of nonconforming product
- Clause 8.3.4: Records of rework
- Clause 8.4: Procedure and records for data analysis
- Clause 8.5.2: Procedure and records for corrective action
- Clause 8.5.3: Procedure and records for preventive action
Take in mind that some of the documents won’t be mandatory if the company doesn’t perform relevant process.
So, QMS will contain a variety of documents, that can be the group into the following (with its respective hierarchy):
- Quality Manual
- Quality Policy
- Procedures and Work Instructions
- Quality Plans and Records
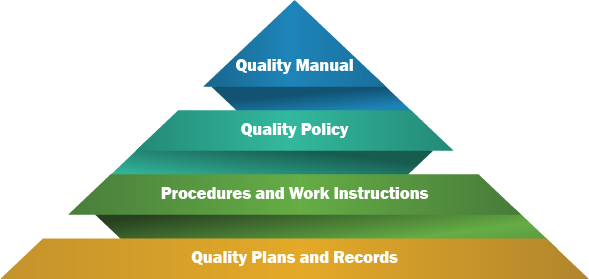
A quality manual is a document that states the company's intentions for operating the processes within the QMS.
A quality policy is a document developed by management to express the directive of the top management with respect to quality.
The term quality procedure refers to the approach used to achieve the goals set by the quality policy.
Work instructions cover the same elements as a procedure but providing greater detail about the activities that should be achieved.
Records provide evidence that a process is in place and performed according to the procedure or work instruction, and that a certain event happened.
A well-structured document system guarantees quality standard is met, reduces the possible errors, reduces downtime when deviations occur due to being able to rapidly access relevant data and allows easy monitoring of the processes. It will improve organizations' efficiency ensuring that its QMS has a strong foundation.